CellCarta's Comprehensive Molecular Profiling Capabilities
CellCarta offers a suite of well-established and emerging molecular platforms such as
- 10X Genomics Chromium and Visium
- digital droplet PCR and much more.
Our laboratories can support the handling and analysis of various specimen types, and our analytical validation process is thorough and supported by partnerships with various reagent manufacturers.
All our workflows, including
- DNA/RNA extraction
- gene expression
- next-generation sequencing (NGS)
- and data analysis
are performed under extensive quality control while keeping your downstream applications in mind.
Using proprietary tools, our team can custom design PCR primers and probes to further support your experiment.
CellCarta accompanies you all the way to data analysis by offering a comprehensive Bio-IT pipeline and adapted molecular analysis software to deliver data analysis solutions that fit your needs.
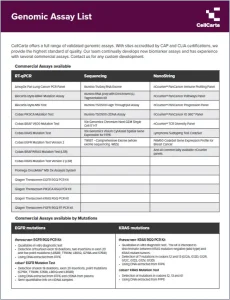
Choose from our extensive list of genomics assays, fit for all phases of research from discovery through clinical trials.
1- DNA/RNA Extraction
Methods used for DNA and RNA extraction can greatly affect downstream analysis.
We have a broad experience with the extraction of nucleic acids from many relevant clinical sample types:
- whole blood, plasma
- cell lines
- FFPE
- FFT
- and fresh tissues
We ensure quality for all of them. We also offer dual extraction possibilities where we simultaneously extract DNA and RNA from the same FFPE material.
Enrichment of specific tissue components or tumor cells by accurate manual microdissection prior to extraction is made possible through the combination of our digital pathology platform and our in-house, board-certified pathologists.
Contact us for more information about our DNA/RNA extraction services!
2- Gene Expression Analysis (qPCR, dPCR)
Gene expression analysis is performed with sensitivity and specificity using PCR-based methods aligned with your study needs.
Our team can support any quantitative PCR-based (qPCR) or digital PCR (dPCR) clinical applications using off-the-shelf assays or validated custom assays.
Learn more about our digital PCR capabilities
Learn more about our quantitative PCR capabilities
3- RNA Profiling
CellCarta provides RNA profiling information using next-generation sequencing (NGS) with the appropriate platforms to tackle any sequencing project size (NovaSeq 6000, NextSeq 550Dx, MiSeq).
For specific gene targets, RT-qPCR and the NanostringTM nCounter® system are offered to accurately quantify custom targets.
Our team also developed a high throughput sequencing method for pre-clinical compound screening with a workflow that can process up to 384 cell lysates without the need for RNA extraction.
Learn more about our RNA sequencing services
4- Mutation Analysis
At CellCarta, DNA mutation detections are performed using well-established single target mutation assays on different PCR platforms (qPCR, dPCR) as well as NGS analysis protocols.
It allows us to detect both single and multiple target mutations while following regulatory requirements. Custom design of sensitive mutation detection assays is also performed.
Harnessing the latest technology (NovaSeq6000, NextSeq 500Dx, MiSeq), our team uses validated panels or generates customized ones, and reports identified variants together with their clinical relevance in a time frame aligned with clinical trials.
In addition to readily available targeted sequencing panels used to detect single nucleotide variants (SNV), small insertions, deletions, copy number variations (CNVs), and tumor mutational burden in specific genes frequently mutated in solid tumors, we also offer comprehensive panels:
- SuperARMS® Pan Lung Cancer PCR Panel by AmoyDx: Detection of 167 mutations and fusion events in 11 target genes specific to lung cancer.
- TruSight Oncology 500 (TSO500) Panel by Illumina: Detection of >500 biomarkers covering multiple solid tumor types and relevant to immuno-oncology, including microsatellite instability (MSI) and tumor mutational burden (TMB) with NovaSeq 6000 and NextSeq 550Dx.
Contact us for more information about our Mutation Analysis services!
5- Fusion Detection
Targeted Next Generation Sequencing (NGS), RNASeq, ARMS PCR, digital PCR, and NanoStringTM nCounter® are all assays we harness for RNA fusion detection.
Validated panels are used to detect cancer specific fusions and point mutations. Some offer the possibility to investigate these fusions without requiring knowledge of their specific break points.
Ready-to-use panels include:
- Archer® FusionPlex Comprehensive Thyroid and Lung (CTL) (NGS): 36 hotspot gene targets in thyroid and lung cancers
- SuperARMS® Pan Lung Cancer PCR Panel by AmoyDx (real-time PCR): 11 target genes for lung samples
- TSO500 Panel by Illumina (NGS): 523 DNA and 55 RNA targets relevant to immuno-oncology, including MSI and TMB.
- NanoStringTM nCounter® – Lymphoma subtyping test (LST)
Customization to your needs:
Panels can be customized to investigate specific fusions and point mutations.
Contact us for more information about our Fusion Detection services!
6- Microsatellite instability (MSI) Assays
Microsatellite instabilities (MSI) result from the systematic accumulation of deletions/insertions in short repetitive DNA sequences in tumor cells due to a deficient mismatch repair (MMR) system.
MSI occurs in approximately 15% of all colorectal cancers and is clinically useful in identifying patients with hereditary nonpolyposis colorectal cancer (HNPCC, Lynch Syndrome) caused by germline mutations of MMR genes.
The MSI status may also predict a patient’s response to certain chemotherapies. More recently, it has been used as a biomarker for immunotherapeutic response, making the MSI status an increasingly relevant tool in genetic and immuno-oncology research.
At CellCarta, we use panels consisting of quasimonomorphic mononucleotide repeats to determine MSI status. These are very sensitive and do not require matching normal tissue or blood.
- The PromegaTM MSI assay v1.2 provides a high throughput solution for multiplex amplification of five repeat markers (BAT-25, BAT-26, NR-21, NR-24 and MONO-27) in the DNA to determine MSI status.
- The IdyllaTM MSI Assay (IVD label) covers seven repeats biomarkers in different genes (ACVR2A, BTBD7, DIDO1, MRE11, RYR3, SEC31A, and SULF2), establishing the MSI status directly from formalin-fixed paraffin embedded (FFPE) tissue in a sample-specific cartridge.
- The TSO500 Panel by Illumina is also used to determine the MSI status.
Contact us for more information about our microsatellite instability services!
7- Single-Cell Profiling and Spatial Gene Expression (10XGenomics – Chromium, Visium)
Using the Chromium system from 10XGenomics, CellCarta can provide single-cell RNASeq profiling.
The heterogeneity of samples normally masked in bulk RNA sequencing analysis can be captured through this method by profiling thousands of individual cells.
The assay is customizable to detect the whole transcriptome, the expression of targeted genes, specific surface proteins, and even single guide RNA (sgRNA) from CRISPR screen.
With the Visium system from 10X Genomics, our team can provide a spatial map of the whole transcriptome within the morphological context of the tissue.
Spatial gene expression data is key in identifying spatiotemporal gene expression patterns.
Contact us for more information about our single-cell and spatial gene expression services